
COMPUTATIONAL DRUG REPROPOSING TO IDENTIFY SARS-COV-2 MPRO INHIBITORS: MOLECULAR DOCKING, ADMET ANALYSIS, AND IN-SILICO/IN-VITRO TOXICITY STUDY
-
 Mohammed Farrag El-Behairy
Department of Organic and Medicinal Chemistry, Faculty of Pharmacy, University of Sadat City, Sadat City, Egypt.
Mohammed Farrag El-Behairy
Department of Organic and Medicinal Chemistry, Faculty of Pharmacy, University of Sadat City, Sadat City, Egypt.
-
Marwa AA Fayed
Department of Pharmacognosy, Faculty of Pharmacy, University of Sadat City, Sadat City, Egypt.
-
Rasha M. Ahmed
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Misr International University, Cairo, Egypt.
-
Inas A. Abdallah
Department of Analytical Chemistry, Faculty of Pharmacy, University of Sadat City, Sadat City, Egypt.
Keywords:
ADMET, Computational chemistry, fragment-based drug discovery, Simeprevir, COVID-19, Structure–activity relationshipsAbstract
Aim and Objective: After the COVID-19 outbreak, drug repurposing has emerged as an effective and fast approach for combating the SARS-CoV-2 crisis. in this work, computational drug repurposing has been utilized to identify new SARS-CoV-2Mpro inhibitors.
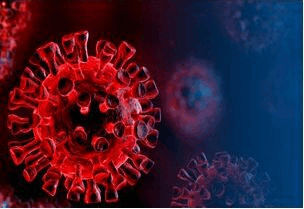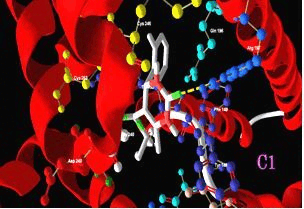
Methods: Comparative molecular docking studies were used to evaluate the activity of the commercially available oral antiviral drug simeprevir and its degradation products (compounds 1–5) against the main protease (Mpro)of SARS-CoV-2 (PDB ID: 6lu7; resolution: 2.16 Å). Moreover, the ADMET and in-silico toxicity properties of the acidic (compounds 1–3) and oxidative (compounds 4 and 5) degradation products of simeprevir were predicted.
Results: Docking studies revealed good binding affinities for compounds (1–5) against Mpro of SARS-CoV-2, with binding free energies ranging from −6.23 to −7.65 kcal/mol. The acidic degradant 2 exhibited the best affinity and was superior to simeprevir and a natural ligand. All compounds were expected to be safe to the CNS.
Conclusion: Compounds 1, 4, and 5 were expected to possess good human intestinal absorption, whereas compounds 2 and 3 appeared to have moderate intestinal absorption.
Peer Review History:
Received: 5 August 2022; Revised: 9 September; Accepted: 20 October; Available online: 15 November 2022
Academic Editor: Dr. DANIYAN Oluwatoyin Michael , Obafemi Awolowo University, ILE-IFE, Nigeria, [email protected]
, Obafemi Awolowo University, ILE-IFE, Nigeria, [email protected]
Reviewers:
Dr. Gehan Fawzy Abdel Raoof Kandeel, Pharmacognosy Department, National Research Centre, Dokki, 12622, Giza, Egypt, [email protected]
Dr. Nicola Micale, University of Messina, Italy, [email protected]
Prof. Cyprian Ogbonna ONYEJI, Obafemi Awolowo University, Ile-Ife, Nigeria, [email protected]
Downloads


Published
How to Cite
Issue
Section

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.







 .
.